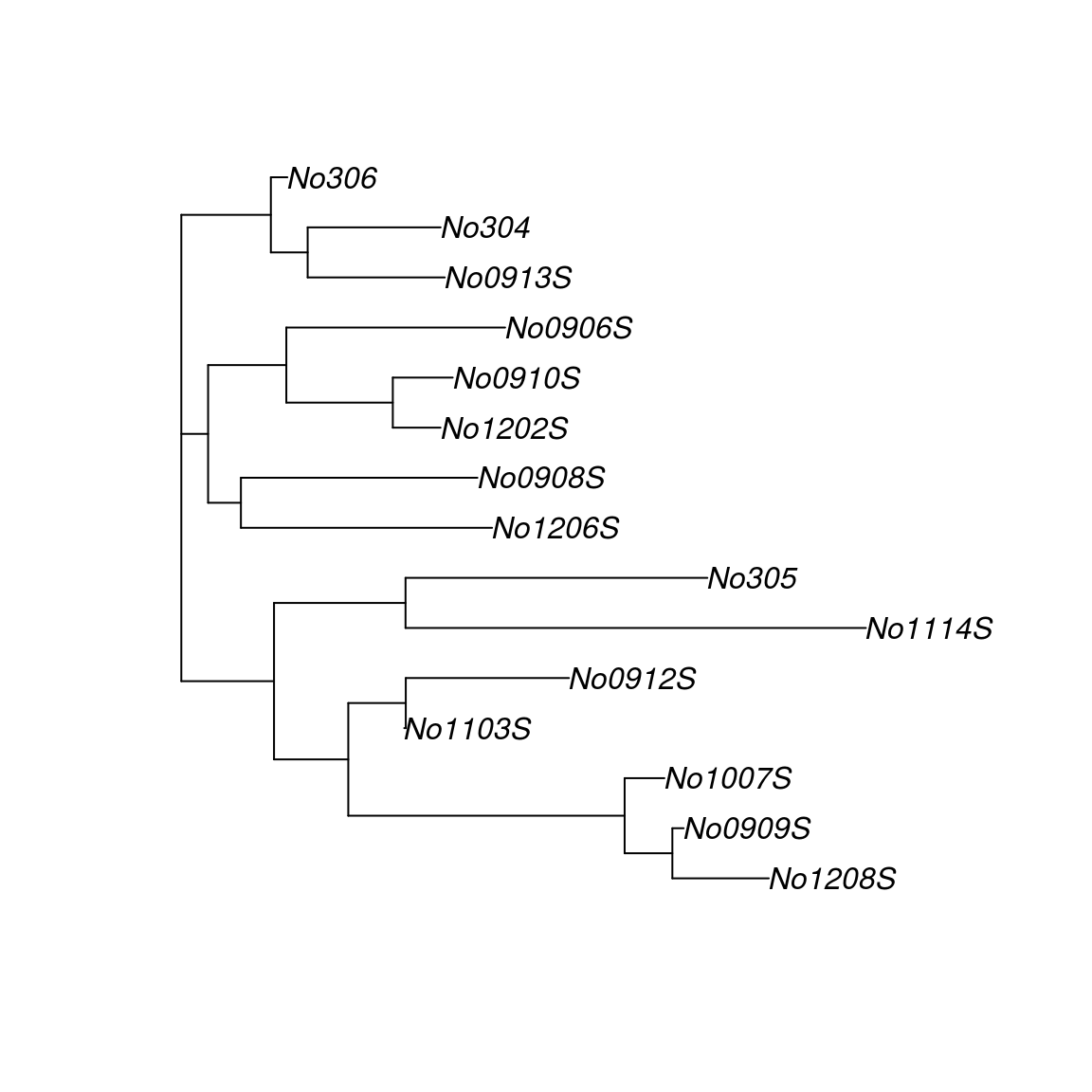
Reading and writing fasta files into R
library(ape)
dna3 <- read.dna("woodmouse.fasta", format = "fasta")
dna3
## 15 DNA sequences in binary format stored in a matrix.
##
## All sequences of same length: 965
##
## Labels:
## No305
## No304
## No306
## No0906S
## No0908S
## No0909S
## ...
##
## Base composition:
## a c g t
## 0.307 0.261 0.126 0.306
class(dna3)
## [1] "DNAbin"
base.freq(dna3)
## a c g t
## 0.3065414 0.2613083 0.1260264 0.3061239
distance = dist.dna(dna3)
class(distance)
## [1] "dist"
length(distance)
## [1] 105
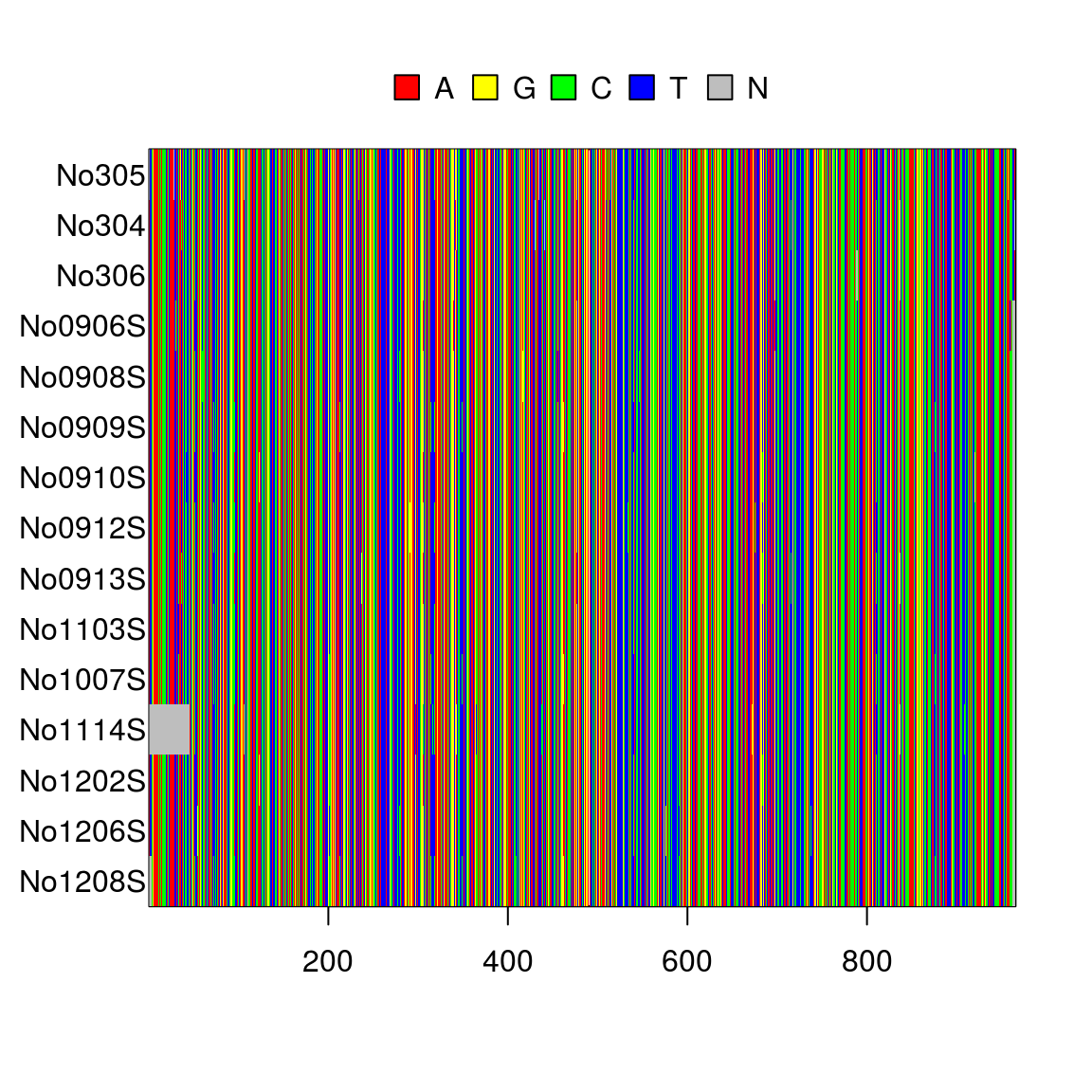
image.DNAbin(dna3)
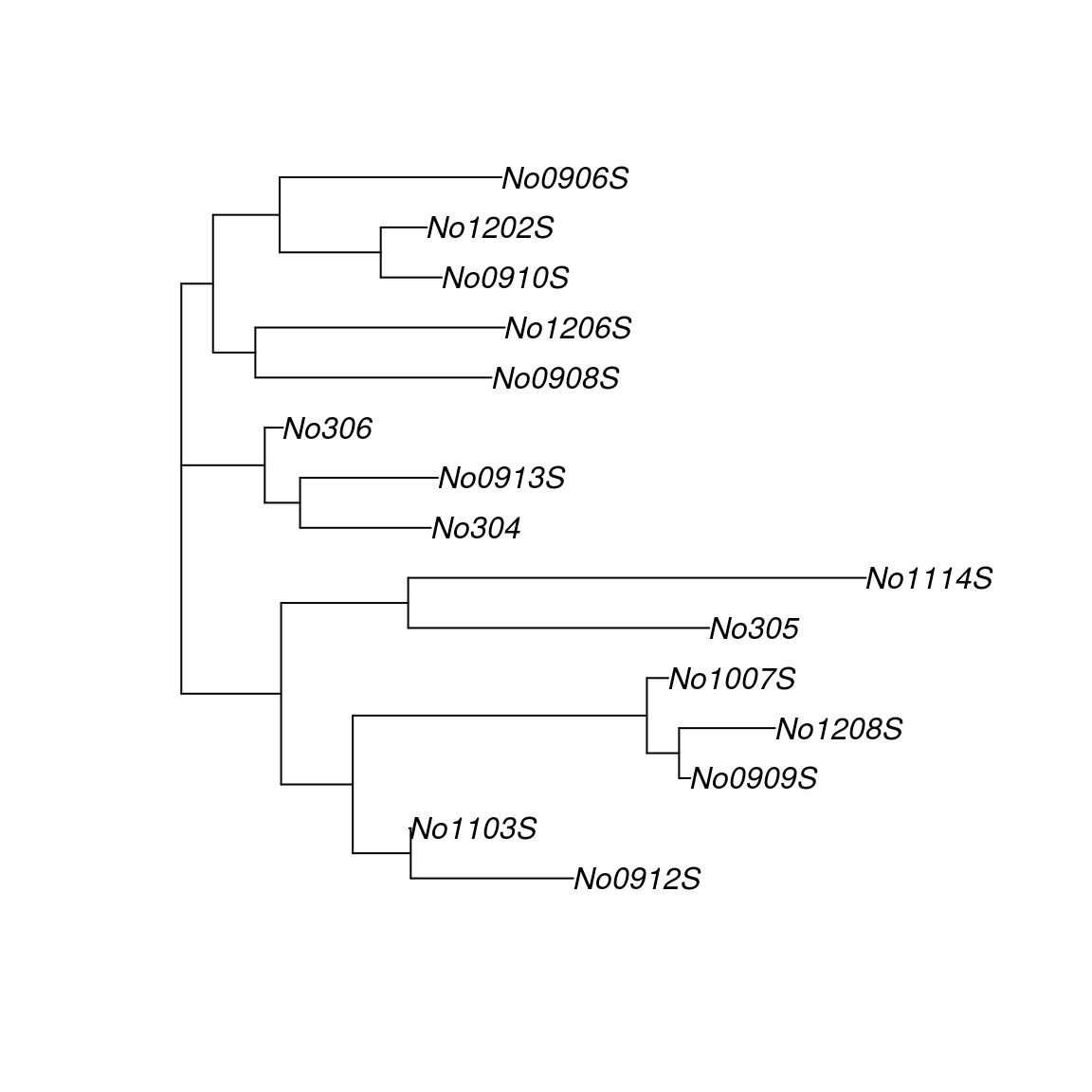
tree.nj <- nj(distance)
class(tree.nj)
## [1] "phylo"
print.phylo(tree.nj)
##
## Phylogenetic tree with 15 tips and 13 internal nodes.
##
## Tip labels:
## No305, No304, No306, No0906S, No0908S, No0909S, ...
##
## Unrooted; includes branch lengths.
write.tree(tree.nj, file = "woodmouse_tree.txt")
file.exists("woodmouse_tree.txt")
## [1] TRUE
plot(tree.nj)
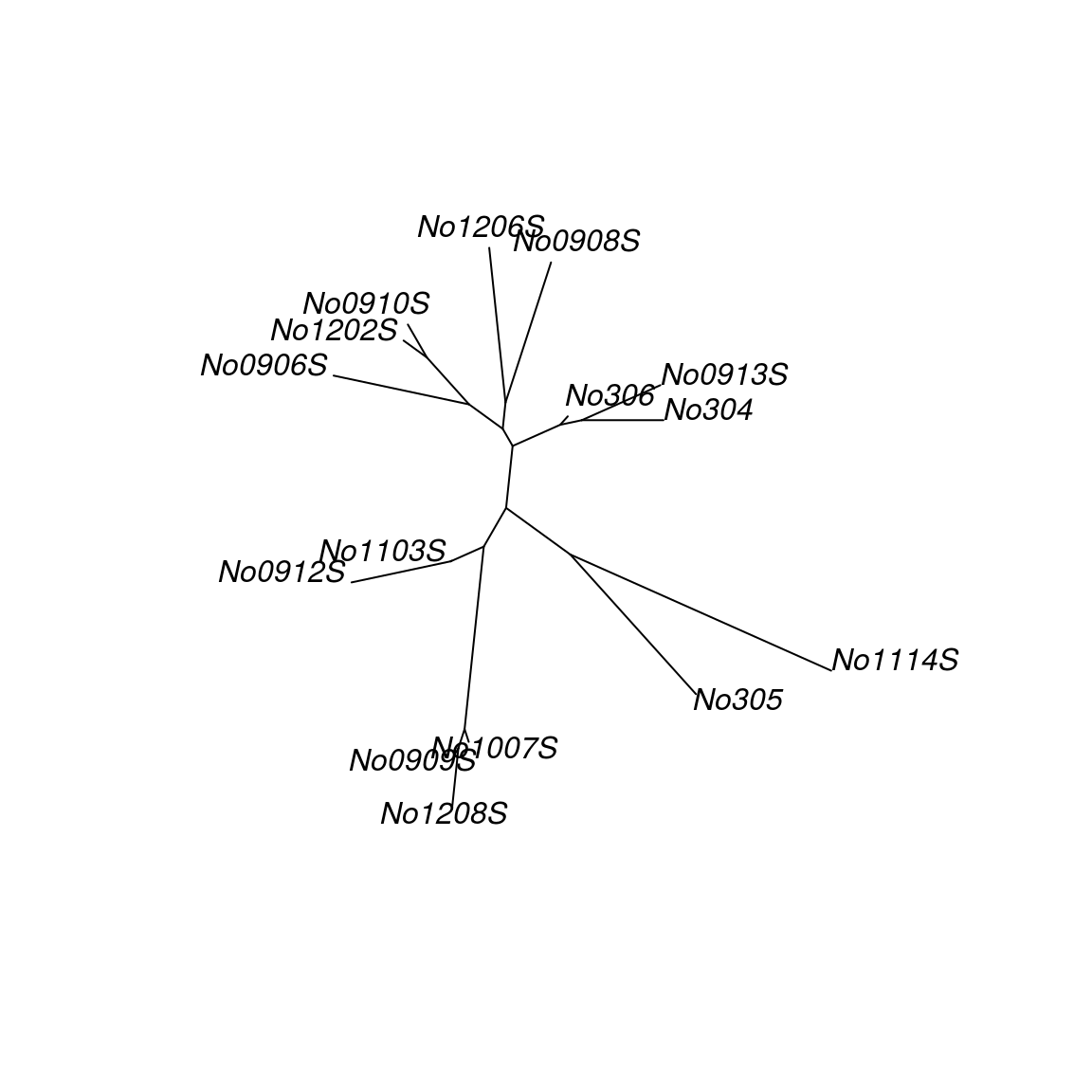
plot(tree.nj, "unrooted")
bio.tree <- bionj(distance)
plot(bio.tree)